Discovery of a new medicine Risdiplam, a Survival of Motor Neuron-2 (SMN2) gene splicing modifier for the treatment of Spinal Muscular Atrophy (SMA)
Targeting RNA drastically expand our target space to therapeutically modulate numerous cellular processes implicated in human diseases. Of particular interest, drugging pre-mRNA splicing appears a very viable strategy, to control levels of splicing product by promoting the inclusion or exclusion of exons.[1] The outstanding progress achieved in this field, will be by highlighted by discussing the breakthrough accomplished recently for the treatment of spinal muscular atrophy (SMA).
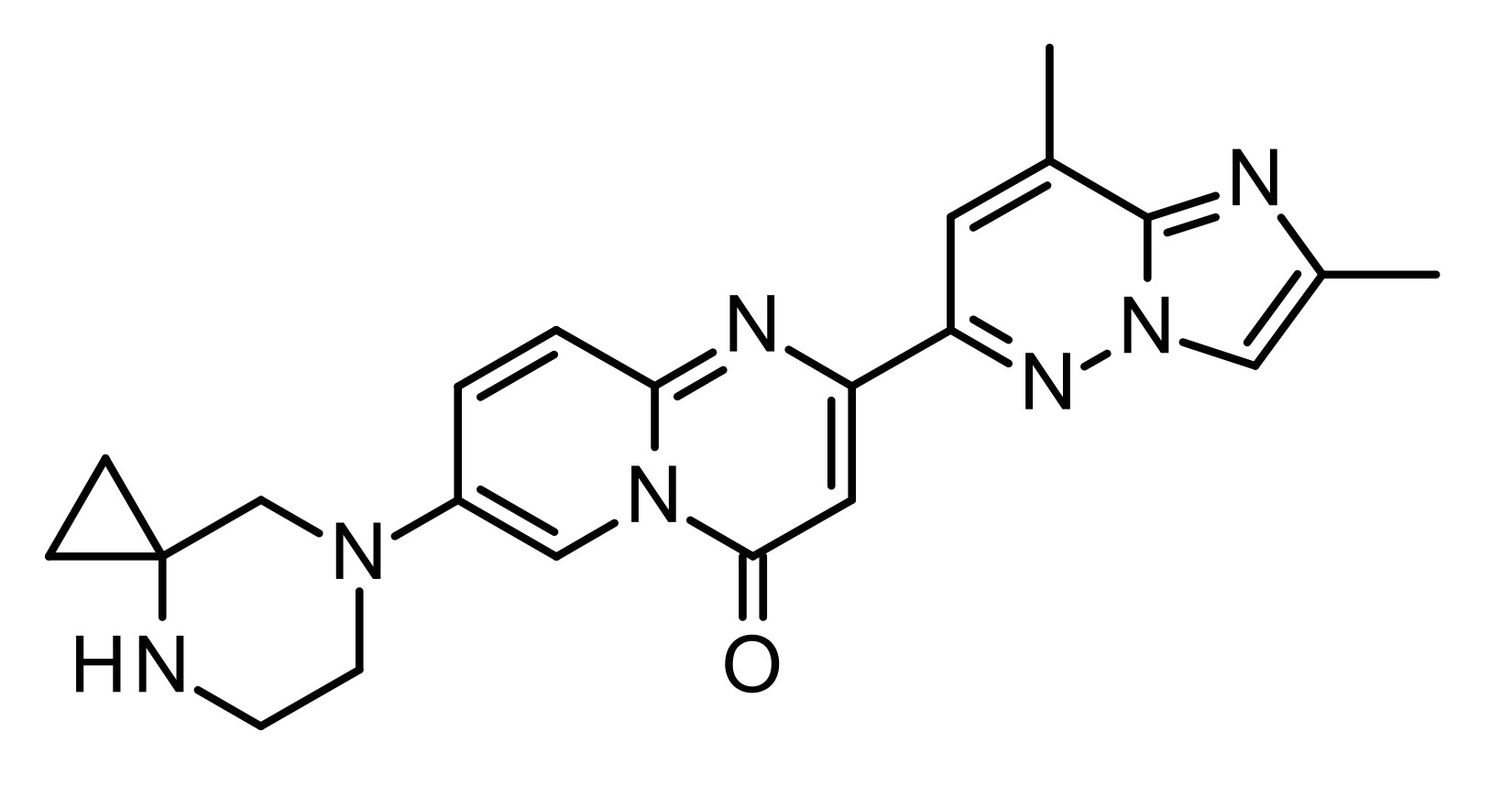
SMA is an inherited disease that leads to loss of motor function and ambulation, and a reduced life expectancy. We have been working to develop orally-administrated, systemically-distributed small molecules to increase levels of functional SMN protein. Our initial development candidate RG7800 was the first SMN2 gene splicing modifier tested in clinical trials in healthy volunteers and SMA patients.[2] It was safe and well tolerated, and increased SMN protein levels up to 2-fold in patients. Nevertheless, its development was stopped as a precautionary measure because retinal toxicity was observed in cynomolgus monkeys after chronic daily oral dosing (39 weeks), at exposures in large excess of those investigated in patients. Herein, we describe the discovery risdiplam (RG7916, RO7034067) that focused on thorough pharmacology, DMPK and safety characterization and optimization.[3] This compound has demonstrated efficacy and safety for the treatment of patients in all ages and stages with SMA, and is now awaiting FDA approval by August 2020.
[1] H. Ratni, M. Ebeling, L. Mueller, Progress in Medicinal Chemistry, 2019, 58, 119-156.
[2] H. Ratni, et al, J. Med. Chem., 2016, 59, 6086-6100.
[3] H. Ratni, et al, J. Med. Chem., 2018, 61, 6501-6517.